
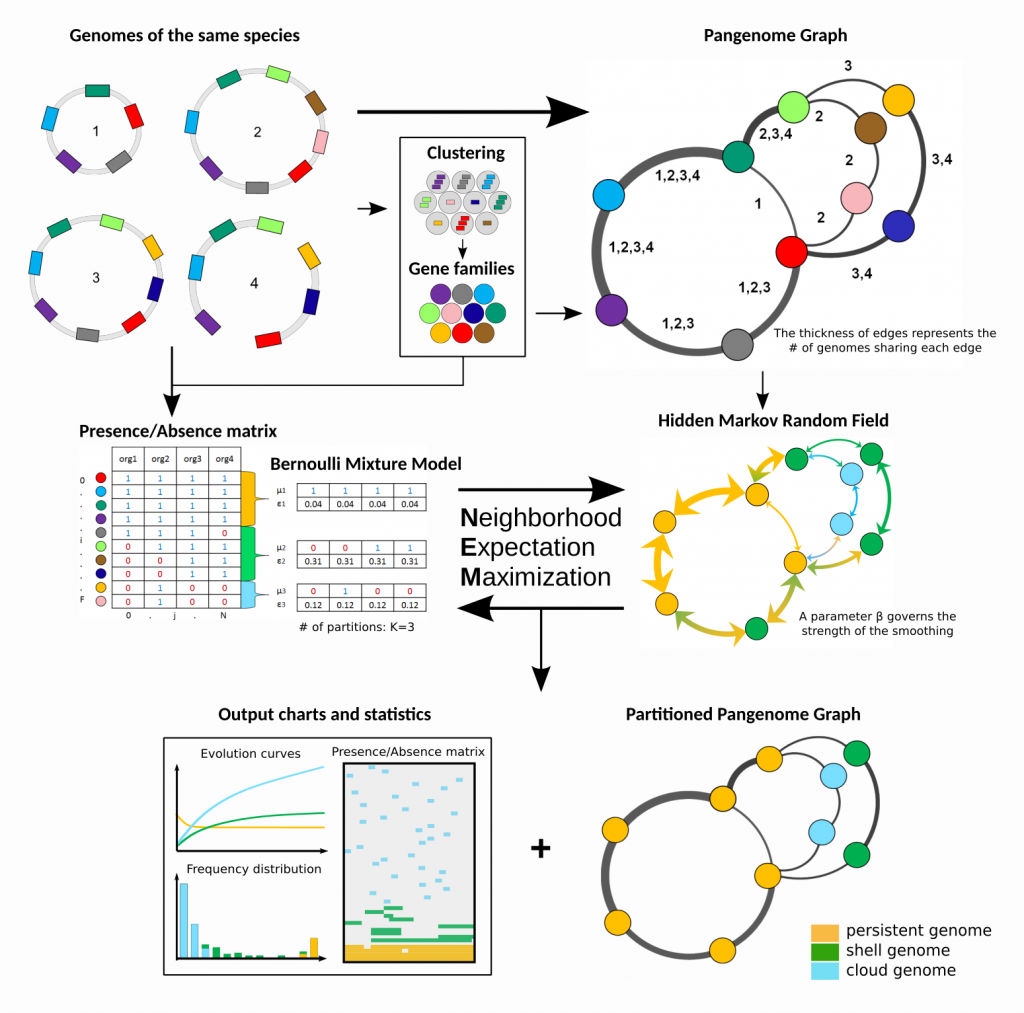
Introduced in microbiology in 2005 by Medini et al. and Tettelin et al., pangenome approaches aim to compile the entire genomic diversity of a species. In these studies, we generally distinguish within the pangenome, the core genome: the set of gene families where gene representatives are present in all organisms; and on the other hand the accessory genome which corresponds to genes specific to certain organisms only. However, we note that the concept of the core genome is limiting with a large number of organisms because genes, although functionally essential, may be absent from the genomes. To limit this phenomenon, almost all studies use an arbitrary threshold of presence (generally 95%) to define a soft core genome. Moreover, this dichotomy between the core and accessory genome does not account for the many frequency ranges at which genes appear in a pangenome. The objective of this thesis work is to propose a statistical approach to partition the pangenome in an original way in order to be resilient to gene absences and to better distinguish between rare and moderately frequent genes. In parallel, several data structures based on pangenome graphs have developed in recent years. Indeed, exploiting all the information available in a genome and no longer just the presence of isolated genes is now crucial to properly account for areas of genomic variability in species. This approach is intended to be the missing link between these new graphic approaches at the sequence scale and the original approaches in isolated gene families. To achieve this, this thesis work therefore focuses on the definition, statistical partitioning and exploitation of a graph of a pangenome as a compact representation of the diversity of the genomic repertoire of a bacterial species.
The major achievement of this thesis is the development of the PPanGGOLiN tool : https://github.com/labgem/PPanGGOLiN
PhD candidate: Guillaume Gautreau – 2016-2020 – https://theses.fr/2020UPASE001
Supervisors: David Vallenet & Claudine Médigue
Publications:
- Bazin A, Gautreau G, Médigue C, Vallenet D, Calteau A. panRGP: a pangenome-based method to predict genomic islands and explore their diversity. Bioinformatics. 2020 Dec 30;36(Suppl_2):i651-i658.
- Gautreau G, Bazin A, Gachet M, Planel R, Burlot L, Dubois M, Perrin A, Médigue C, Calteau A, Cruveiller S, Matias C, Ambroise C, Rocha EPC, Vallenet D. PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph. PLoS Comput Biol. 2020 Mar 19;16(3):e1007732.
- Vallenet D, Calteau A, Dubois M, Amours P, Bazin A, Beuvin M, Burlot L, Bussell X, Fouteau S, Gautreau G, Lajus A, Langlois J, Planel R, Roche D, Rollin J, Rouy Z, Sabatet V, Médigue C. MicroScope: an integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucleic Acids Res. 2020 Jan 8;48(D1):D579-D589.